These guidelines and procedures have been identified as being beneficial in the various REB application and reporting processes.
Travel Service Providers
Guidelines for Electronic Written Informed Consent (eIC)
Definition Of Secondary Analyses Of Clinical Data
The secondary use of data refers to the use in research of clinical data already contained in participant’s health record and collected through the normal provision of routine care.
The Provincial Personal Health Information Protection Act of Ontario (PHIPA) sets forth the following requirements for the secondary use of clinical data in research:
- The scientific question being asked is valuable and justifiable;
- Appropriate measures are being taken to protect the privacy of the individuals, to ensure the confidentiality of the data, and to minimize harm to participantss;
- The patients regarding whom the data refer will not be directly contacted by the researchers without their prior express and informed consent;
- Identifying information will not be released to researchers outside the hospital without the patient’s express and informed consent;
- Individuals to whom the data refers have not previously objected to such secondary use;
- The data being collected would normally be obtained in the provision of care (section 29 (2)). As a consequence, the REB must ensure that the research plan limits its analysis to those data fields that would normally be available in the health record (departmental or centralized chart). If an investigator wishes to look at variables that are supplemental to care (e.g., in many instances, this could include race and SES), the individual’s express and informed consent must first be obtained and the research plan approved by the board.
- It should be noted that PHIPA (health privacy legislation in Ontario) allows researchers to use clinical data to answer questions on the basis of ‘implied consent’. Implied consent can be assumed if the institution has posted public notices to advise patients of the different uses that it makes of personal health information & that an REB has approved the research plan. CHEO has fully implemented the legislation and posted notices are found in high traffic areas around the hospital.
Pediatric Research Blood Volume Draw Guidance
The table below sets out guidance for researchers and REB reviewers for assessing the risk level associated with total research blood volumes (TBV) drawn from children and adolescent research participants. Clinical blood work must always take precedence over research blood work. When research blood volumes are large, the most responsible physician should ensure that the patient’s clinical needs are met first and foremost. In addition, the investigators should make every effort to minimize the discomfort associated with blood draws.
- • Research blood volumes falling within the limits outlined below may be considered minimal risk. Additionally, maximal blood draws (5% of TBV monthly) should not occur more than 3 months in a row.
- • Research blood volumes falling above the volume indicated in the table below will be required to undergo Full Board review. Justification for these research blood volumes should be described in the REB application, and the study protocol.
- • Research blood draws in infants with a body weight of less than 7.3 kg (16 lbs) and/or less than 6 months of age will always be reviewed at the Full Board.
Translated Documents
Informed consent forms must normally be available in both English and French. This requirement is based on the importance of free and informed consent in research, and the hospital’s commitment to bilingual service for families and youth.
Under limited circumstances, the REB can waive the translation requirement. In order to obtain such a waiver, the investigator must demonstrate that it is either inappropriate or impracticable to require both French and English consent forms. In other words, the investigator would need to show that the additional financial, material, human, organizational and other resources needed to carry out a study in both languages are so burdensome as to render the research unfeasible. An investigator might argue, for example, that a study cannot be responsibly conducted in one or other official language because the outcome measures have not been yet been translated and the resources needed to do so are prohibitive.
The decision to waive the requirement for consent forms in both official languages will be sensitive to the specific nature of the study. Projects are not likely to be given a waiver if they present more than minimal risk to subjects or if they offer innovative therapies to patients that would otherwise be unavailable.
There are several options open to investigators who wish to have a document translated into French. For funded projects, translation services have been negotiated with Mrs. Rose Gorrie at [email protected]. All requests for translation are to include a completed Request for Translation form which is available on CHEOnet. The REB will also accept documents translated by person that has an A+ level (or a reasonable equivalent).
Investigators must ensure that the French and English versions of the consent form are equivalent in every aspect. In order to do so, a professional translator should normally carry out the work.
Research studies requiring French translation cannot be approved until the French documentation has been submitted and approved, or confirmation provided to the REB that the documentation has been submitted for translation (a copy of the email or letter to the translator).
This policy requires that a French (or English) version of the consent form be made available within two to three months of the initial approval. However, if an investigator believes that it is impossible to meet this time line, the Board should be advised and a reasonable alternate time line proposed.
Guidance on Case Report
Case reports requiring REB review
Scope
The purpose of this section is to provide guidance on when the publication/presentation of case reports and/or case series constitutes human subjects research that requires REB review and approval. It is common to report unique and interesting clinical cases through publication in medical journals or presentation at medical or scientific meetings. This gives rise to questions regarding when a case report or case series becomes research requiring REB review and approval.
What is the distinction between a case report and a case series?
Case report: A detailed report on an individual patient (e.g., presentation, diagnosis, treatment, response and follow-up) arising from routine clinical practice. The report describes the course of medical treatment of one or more patients that has a unique outcome or the handling of a unique clinical case. Consent for publication is required by the patient for case reports.
Case series: A presentation of information or a collection of case reports on more than three patients.
When is REB review and approval required?
A case report or a case series requires REB review and approval when it meets the definition of research as “an undertaking intended to extend knowledge through a disciplined inquiry or systematic investigation.”
A case report with three or fewer patients does not require REB review and approval. This is because it generally does not meet the definition of research on the basis that there is:
- A limited number of ‘unique’ patient/cases from a single physician or clinic (≤3)
- No research intent at the time of the intervention (i.e., no prospective plan to systematically evaluate the outcomes for purposes other than treatment)
- No systematic data collection (e.g., chart reviews)
- No intention to test or compare various therapies/outcomes/data
- No descriptive or comparison statistics between cases
- Not considered to be generalizable information
Conversely, if anything was done in the course of treatment with a research intent or that aims to answer a specific research question, the case report becomes research that requires REB review and approval.
A case report with more than three patients becomes a case series that is considered research because it necessarily meets at least one of the criteria above and requires REB review and approval.
In accordance with TCPS2 Article 6.11, researchers must submit their application prior to the start of any research activity. REBs do not grant “retroactive” approval after research is complete. Clinicians/researchers are advised to consult with the REB when there is uncertainty as to whether an activity constitutes human subjects research that requires REB review and approval.
Guidance on Types of Data
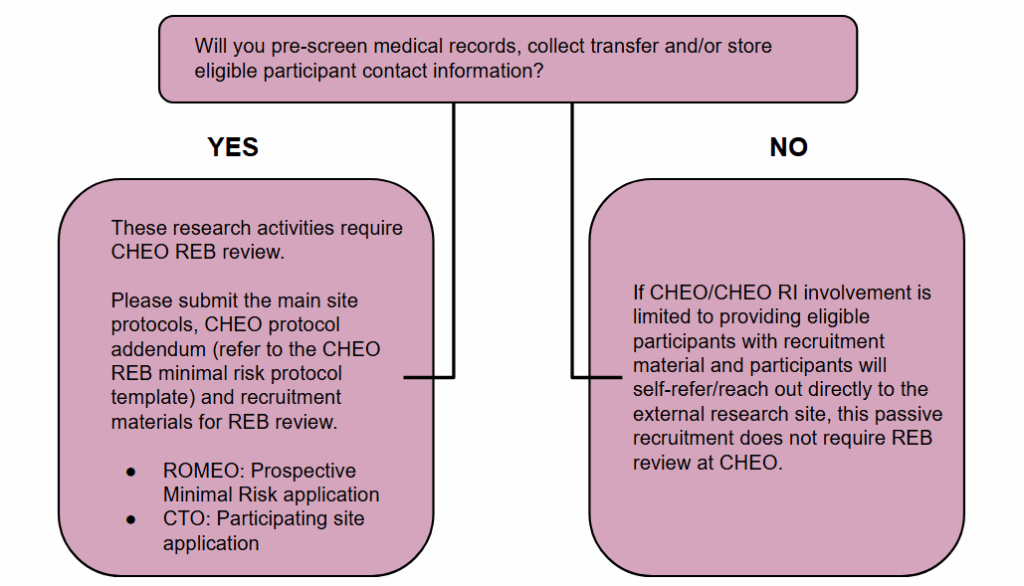
Passive Recruitment
Passive recruitment for research conducted at external institutions entails informing potential participants about studies taking place outside of CHEO/CHEO RI, without any direct involvement from CHEO/CHEO RI. This process is limited to providing general information, whereby interested individuals independently initiate contact with the external research teams. Importantly, there is no exchange, collection, or transfer of personal or study-related data between CHEO/CHEO RI and the external sites.
Passive recruitment in research involves strategies where potential participants are provided recruitment materials (e.g., posters, information handouts). Of note, if any of the study will be explained by the research team at CHEO/CHEO RI this would not constitute passive recruitment.
Where research activities at CHEO are limited to passive recruitment, this activity may not require CHEO REB review and approval.